

生命現象の変化を情報科学技術で捉える
生命は地球上の至る所に存在し、生命が存在しない場所を想像するのは困難です。それを支える分子的なシステムは多様であり、それらを深く理解して得られた知見を人類社会に還元するには、多くのアプローチが必要です。当研究室は情報科学を用いてオミクスデータなど大規模データから変化を捉え、新しい発見に繋げたり、あるいは新しい技術の開発を目指しています。
複雑なゲノムの配列解析

昨今のゲノムシーケンシング技術の発展は目覚ましく、ゲノム配列の解析も対象を拡げています。植物はゲノムが複雑なものが多いですが、精度の高いロングリードの登場により扱いやすくなってきました。データ解析を通じて新たな知見の獲得やその基盤作りを目指して研究しています。

ヒトを始め多くの真核生物(細胞内に核を持つ生物)は両親から受け継いだ2セットの染色体を持ち2倍体ゲノムと呼ばれています。母方由来の染色体と父方由来の染色体に当たります。それぞれの相同染色体は程度の違いはありますが、一般に異なる対立遺伝子を持ち、これらの新しい組み合わせで新世代の生命が誕生することになります。

バイオインフォマティクスでこういったゲノム配列を扱う際に気をつけなければいけない事は、我々はシーケンシングデータの由来染色体が分からないという事です。最近は相同染色体を実験的に分ける方法も出てきましたが、大抵のケースで由来不明のバラバラの配列が、しかも大量に出てきます。2倍体の場合似たような配列が2セット分(母方由来のデータと父方由来のデータ)出てきますが、これが混ざってしまうと色々と面倒なことになります。このため、例えば2倍体では母方と父方のデータを計算で分けることが重要になります。とは言えこれも難しく、つい最近までは分別することを諦めて、平均的な描像に焦点を当てる時代が続いてきました。
ところが、農学上有用な植物や一部の生物はこれを2セットではなく4セットや8セットも保持しており4 倍体や8倍体、総称して多倍体と呼ばれています。似たような配列が2セットあるだけでも厄介なのですが、これがさらに倍、その倍の倍となると何が起こるかは何となくお分かりいただけるのではないかと思います。混ざってしまい大変扱いにくい対象となります。
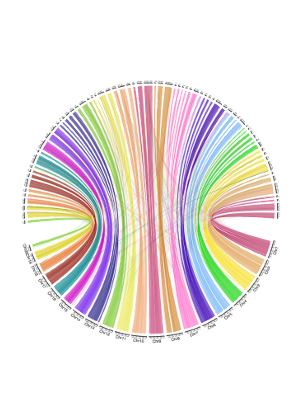
左図は異質4倍体でかつ比較的巨大なゲノムであるA.hypogaea(ラッカセイ)のアセンブリを示したものです。上半分が我々の結果で、下半分が大掛かりなプロジェクトで再構築された参照ゲノムとなり、それぞれが異なる色の線で結ばれて対応関係を表しています。完全とはいきませんが、下半分の各染色体が上半分の3-4配列(コンティグといいます)でカバーされており、2週間程度で行った実験と解析としては驚きました。ラッカセイのゲノムはお互いに似ているサブゲノムAとサブゲノムBがあり、とても混ざりやすいデータでした。これらを計算機がきちんと識別出来たことが重要だと考えています。

上の実験は2020年の終わり頃に出てきた新技術をベースにしており、長く正確に読めることが特徴です。DNAやRNAを長く正確に読めるのは、このように大変重要です。各々の由来する染色体に応じてデータを見分けやすくなるからです。数年前まで諦めていたようなプロジェクトが現実的になってきており、より深く分子の挙動や変化を捉えるためバイオインフォマティクスの観点からこの新しい分野で研究を進めています。
寄生と進化

自然界には実に多様な寄生形態が存在しています。寄生する生物の進化はユニークなことが多く、ミトコンドリアといった細胞小器官レベルで影響が観測されるケースもあります。動植物へのウイルスや細菌感染、そして共生も寄生という側面があると考えています。寄生を生物が行うとき、分子レベルで何が起こったのか、起こりうるのかを研究しています。
進化の過程で生命は多様な環境に適応してきました。多くの状況に備えるため、いわば生き残りの道具となる遺伝子を多く保持する方が有利に思えます。ただ、これにはおそらくエネルギー的な限界があり、遺伝子を無限に保持出来るわけではありません。キャンプに行く時、アーミーナイフは大変便利ですが、それにしたところで入れられる道具の数には限度があります。しかも重くなります。やはり、道具は軽い方が良いです。

ある種の生命は他の生命にエネルギー的に依存するという形態を取ることがあり、寄生体と呼ばれます。腸にいる回虫、赤痢アメーバやトリパノソーマといった病原体、もっと小さくなると寄生性の細菌もあります。前職でサウジアラビアにいる時、寄生性の植物もいると知りより興味を持ちました。こういった生命の特徴は独立して生存することを部分的に、あるいは完全に放棄している点です。興味深いことに、寄生性の生命は遺伝子を「落とす」傾向があることです。要らないので捨ててしまう、より正確にはある時点で必要だった遺伝子を失った祖先すら生存できた、と言えるでしょうか。寄生先から拝借するわけで、こういった進化は縮退進化と呼ばれています。やはり道具は軽い方が良いわけで、最終的にはシンプルなナイフになるのでしょうか。

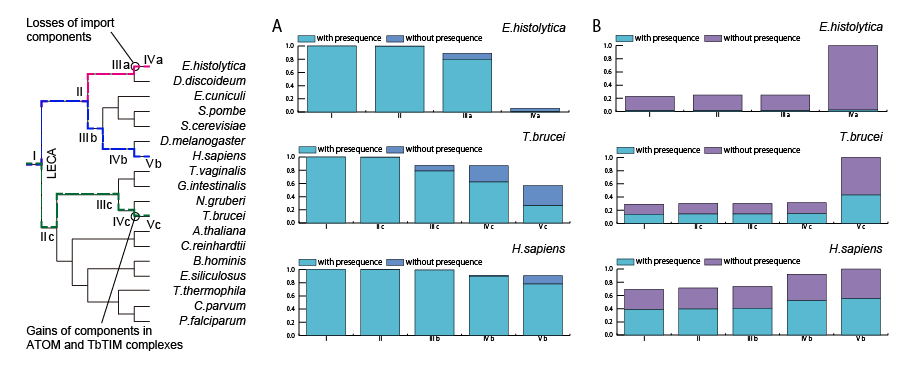
単純にそうではないと考えています。上図のA列では寄生性の生物がいかに遺伝子を失っているかを示しており、対比として一番下にヒトを置いています。左から右に時間の流れを示しています。B列は逆にある細胞小器官にある遺伝子がいつ獲得されたかの推定時期を示しています。下段のヒトの場合は遺伝子の消失や獲得がなだらかですが、上段は非常に極端であり、中段の生物は中間といったところだと考えています。上段と中段の生物は寄生性の原生生物ですが、B列で示すように獲得も環境が変化するタイミングで行なっていると分かり、やはり環境に応じて遺伝子セットの入れ替えが必須なのだと理解しました。消失の説明に比べ、どうやって新規遺伝子を獲得するのかは説明が難しいと感じます。この意味でもゲノムの複製や多倍体ゲノムの研究は重要です。
統合解析

ゲノムやトランスリプトームなどの同じ性質のデータだけではなく、異なる性質のデータを組み合わせて現象を説明する統合解析が重要になってきています。非常に未成熟な分野ですが、まずは人が理解するためのアノテーションの文脈で特に重要だと考えており、研究に励んでいます。

ここ数年でインフォマティクスが扱える範囲が飛躍的に、本当に驚くほどに広がりを見せています。2022年の後半からChatGPTが世界中で話題になっていますが、LLMだけではありません。Multimodal解析といわれる分野に興味を持っています。例えば、DALL-E(2)のベースとなるCLIPというものがありますが、自然言語と画像を同時に学習する凄さを見せつけられている思いです。Multimodalな学習を可能にしたのは、画像とキャプションのペアが大量に得られたことが一つ重要であり、大規模なデータの組み合わせは興味が尽きません。
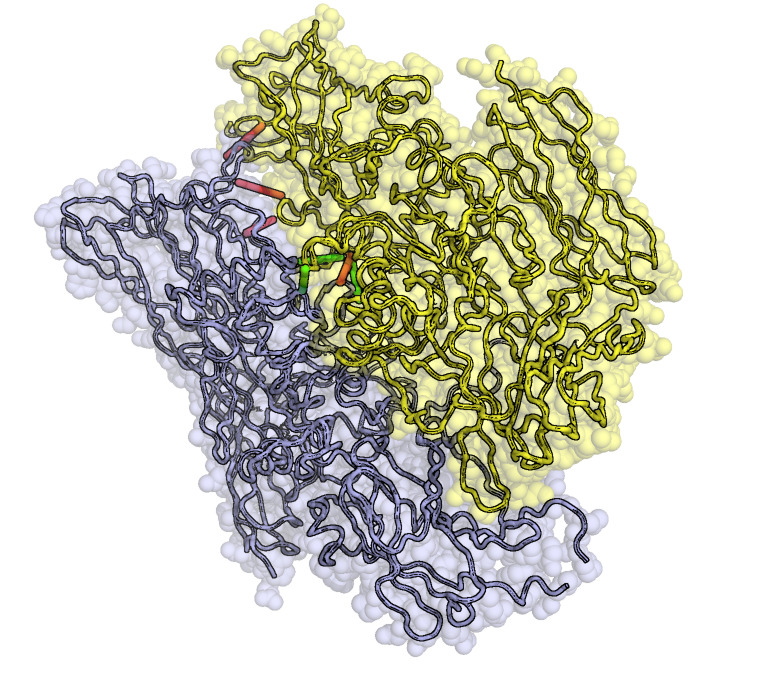
異なった種類のデータから情報を抽出し、計算としてまとめるのはやりがいのある仕事です。分子の立体構造を決める方法の1つに、X線結晶構造解析と呼ばれる手法がありよく使われてきました。データとしては各アミノ酸残基を構成する原子(炭素原子や窒素原子など)の3次元座標が並んでいる形式のファイルを扱うのですが、解析する際に他から情報を持ってきても良いわけです。残基間距離を大量の配列情報から予測することが可能と当時分かってきたので、これを利用しました(コンタクト予測と呼ばれています)。結晶中の3次元情報と、大量の遺伝情報から計算される残基間距離を使って、結晶内で重要な相互作用面を判別する手法を開発しました。ゲノム上の変異は、少なくともタンパク質を記述する遺伝子においてはランダムに蓄積しておらず、物理化学的な制約を非常に強く受けることが背景にあります。大規模なゲノム情報と立体構造情報はこうして繋げられるわけで、自然の奥深さを感じます。
こういった大規模データの組み合わせは、より工学寄りの研究になりますが、応用に関して頭を柔らかくしながら、研究していきたいと思います。
A monkey is a machine that preserves genes up trees, a fish is a machine that preserves genes in the water; there is even a small worm that preserves genes in German beer mats. DNA works in mysterious ways.
Richard Dawkins, The Selfish Gene